Using IM-UFF
-
I am hoping to simulate the UiO-66 mof with cadmium. I just found out about IM-UFF and I am hoping to simulate my system with this reactive force field (to show chemical adsorption).
But from the 2017 paper (doi.org/10.1016/j.jmgm.2017.08.023) on this force field, it was mentioned that the use of IM-UFF is recommended on "moderately sized systems (up to 100 molecules)".
Is it possible to simulate larger systems (say 2000 to 3000 atoms) by not displaying system updates in the GUI of Samson? By that I mean is it possible to run a simulation using only the Python console?With these questions, I am also wondering, can IM-UFF be used in non-interactive simulations? I am hoping to only take advantage of it being a reactive force field. I appreciate any help on this matter.
Thank you!
-
Dear @Joseph-1 ,
The mentioned limitation was mainly for the performance on average laptops some years ago. You can try using UFF/IM-UFF on bigger systems - I used it on systems with thousands and tens of thousands of atoms.
It is possible to launch the simulation using Python, but to improve the performance, you can simply increase the number of steps with which the viewport is updated in the chosen State Updater - this should improve the speed.
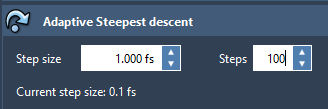 (here the positions of the system in the viewport will be updated every 100 steps).
(here the positions of the system in the viewport will be updated every 100 steps).You can use IM-UFF for non-interactive simulations as well, or simply try using UFF (IM-UFF is an advanced and interactive version of UFF).
Please note, that, for now, it does not support periodic boundary conditions. But if you want you can freeze atoms at the boundaries, see for example:
- https://blog.samson-connect.net/freezing-atoms-positions
- User guide: Minimizing a part of a molecule (the Freeze steps be applied to the simulation as well).
Please let us know if you have any questions.
-
I highly appreciate the response. Thank you for the information!
Is it possible to use LAMMPs within the SAMSON software? Can LAMMPs implement the periodic boundary conditions and use IM-UFF within SAMSON?
-
Currently, there is no public extension in SAMSON that can launch/use LAMMPS. But you can export your system into a LAMMPS Data format and load LAMMPS files (data and trajectories) back in SAMSON.
We plan on adding the support for periodic boundary conditions in one of the future releases of SAMSON. For some systems, you can simulate without PBC or by freezing the boundary atoms to fix them.
You can also combine simulations in SAMSON with e.g. ASE package using Python, see Python scripting: Combining SAMSON simulation with ASE calculators