Create GMX data file, problem with GROMACS Model Generator
-
Dear SAMSON Team!
I would like to run a simple simulation in Gromacs with some water molecules.
I drew molecule in Avogadro, and create 10 molecules with Packmol and saved as pdb file.
I load this file in SAMSON and connect atoms with Add Simulator->Untick Use existing bonds.
I tried to generate GROMACS data file with GROMACS Model Generator, chose OPLS-AA force field and ran. It could generate easily gro files, but whenever I try to generate pdb files, it crashes. I tried to choose other force fields but the result was same.
What did I do wrong?
I attached the file (in py format).
Thank you in advance for your help!
Best regards,
Patrik Molnár0_1586588617680_watersys.py -
Sorry, I forgot another question: If I generate this data file, will it contain all necessary information about my system (and I need to scripts just an input file)?
-
Dear @Patrik ,
Thank you for reporting this issue. I can reproduce it and will try to fix it.
If I generate this data file, will it contain all necessary information about my system
Yes, you can launch GROMACS simulations with it.
But you may launch them also with GROMACS Wizard, see GROMACS Wizard tutorial.
By the way, if you want to manually create a water system you may very easily do it in SAMSON thanks to new Add editor:
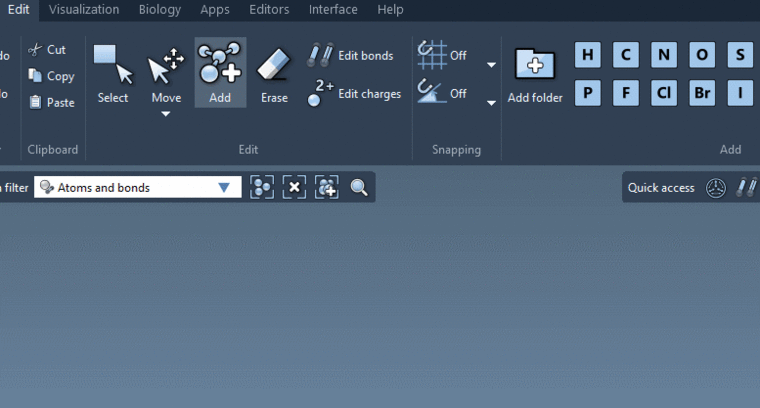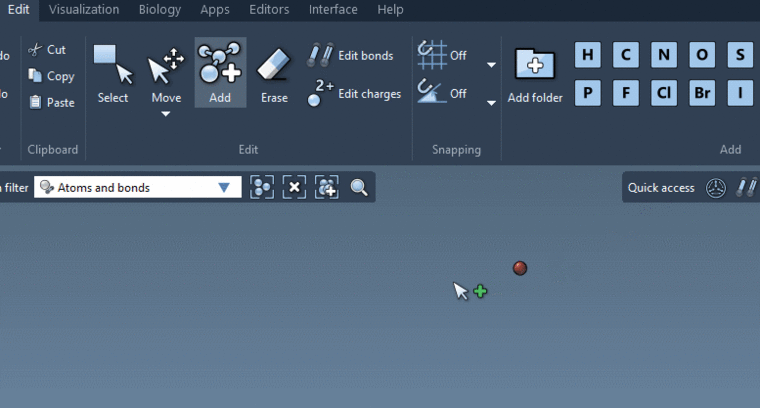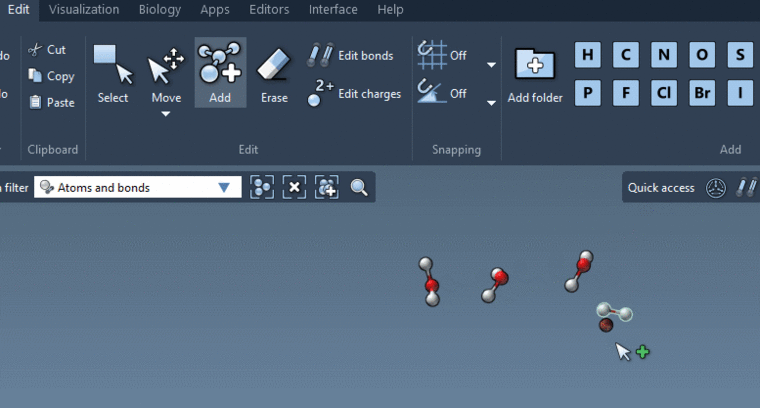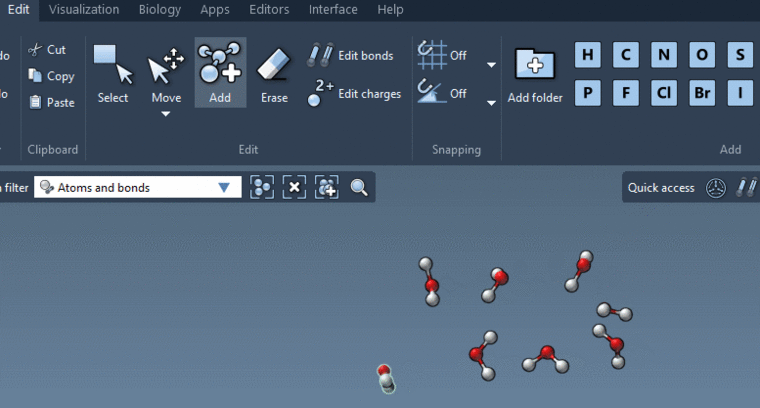
For more information, see What’s new in SAMSON 2020. -
Dear @DmitriyMarin
Awfully thank you for your reply!
"Yes, you can launch GROMACS simulations with it." -> Does it mean also that it is not necessary to add Gromacs Wizard and I can use any version of Gromacs?
Thank you for the trick about Add editor.Best regards,
Patrik Molnár -
@patrik said in Create GMX data file, problem with GROMACS Model Generator:
"Yes, you can launch GROMACS simulations with it." -> Does it mean also that it is not necessary to add Gromacs Wizard and I can use any version of Gromacs?
Yes, you can launch simulation using another version of Gromacs but outside of SAMSON.
I have updated the PDB Importer, so now your water molecules will be covalently linked on the import so you do not need to apply a simulator with UFF.
-
@dmitriymarin Thank you
-
@Patrik ,
We have fixed the reported issue with the Gromacs Model Generator. It should be automatically updated for you once you relaunch SAMSON.
-
Yes, I could already run it.
Thank you!