Fatal error trying to use Gromacs Wizard : gro file does not have the number of atoms on the second line
-
Hello, trying to use the Gromacs Wizard to prepare a simulation based, on the tutorial founded here http://www.mdtutorials.com/gmx/lysozyme/04_ions.html. Trying to make the preparation step, generating the box, adding solvent and ions. When I click on prepare I nearly immediately got a fatal error :
*Program: gmx editconf, version 2020.3-MODIFIED
Source file: src\gromacs\fileio\groio.cpp (line 67)Fatal error:
gro file does not have the number of atoms on the second line*Finding the gro file in my working directory I can find that its weight is 0 ko, and opening it with notepad, there's nothing written in.
I try to generate a gro file with gromacs model generator and use it to replace the gro file of the working directory, but this doesn't work.
Any idea on how to solve this ?
Here the output file :
######################################## PREPARATION STARTED ########################################
:-) GROMACS - gmx pdb2gmx, 2020.3-MODIFIED (-: GROMACS is written by:Emile Apol Rossen Apostolov Paul Bauer Herman J.C. Berendsen
Par Bjelkmar Christian Blau Viacheslav Bolnykh Kevin Boyd
Aldert van Buuren Rudi van Drunen Anton Feenstra Alan Gray
Gerrit Groenhof Anca Hamuraru Vincent Hindriksen M. Eric Irrgang
Aleksei Iupinov Christoph Junghans Joe Jordan Dimitrios Karkoulis
Peter Kasson Jiri Kraus Carsten Kutzner Per Larsson
Justin A. Lemkul Viveca Lindahl Magnus Lundborg Erik Marklund
Pascal Merz Pieter Meulenhoff Teemu Murtola Szilard Pall
Sander Pronk Roland Schulz Michael Shirts Alexey Shvetsov
Alfons Sijbers Peter Tieleman Jon Vincent Teemu Virolainen
Christian Wennberg Maarten Wolf Artem Zhmurov
and the project leaders:
Mark Abraham, Berk Hess, Erik Lindahl, and David van der SpoelCopyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2019, The GROMACS development team at
Uppsala University, Stockholm University and
the Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.GROMACS is free software; you can redistribute it and/or modify it
under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.GROMACS: gmx pdb2gmx, version 2020.3-MODIFIED
Executable: C:\Users\User\AppData\Local\OneAngstrom\SAMSON-Data\1.0.0\Scratch\Elements\02407D21-0490-30BA-D20F-2E88DB100FC5\Resource\gromacs\gmx.exe
Data prefix: C:\Program Files\Gromacs
Working dir: C:\Users\User\AppData\Local\OneAngstrom\SAMSON-Data\1.0.0\Scratch\Elements\02407D21-0490-30BA-D20F-2E88DB100FC5\Resource\gromacs
Command line:
gmx pdb2gmx -f C:/Users/User/AppData/Local/OneAngstrom/SAMSON-Data/1.0.0/Scratch/GROMACS/PDB/temp.pdb -o C:/Users/User/AppData/Local/OneAngstrom/SAMSON-Data/1.0.0/Scratch/GROMACS/GeneratedFiles/1AKI_model.gro -i C:/Users/User/AppData/Local/OneAngstrom/SAMSON-Data/1.0.0/Scratch/GROMACS/GeneratedFiles/1AKI_model.itp -ff oplsaa -water spc -p C:/Users/User/AppData/Local/OneAngstrom/SAMSON-Data/1.0.0/Scratch/GROMACS/GeneratedFiles/1AKI_model.top -ignhOpening force field file ./oplsaa.ff/aminoacids.r2b
All occupancy fields zero. This is probably not an X-Ray structure
Opening force field file ./oplsaa.ff/atomtypes.atp
Opening force field file ./oplsaa.ff/aminoacids.rtp
Opening force field file ./oplsaa.ff/aminoacids.hdb
Opening force field file ./oplsaa.ff/aminoacids.n.tdb
Opening force field file ./oplsaa.ff/aminoacids.c.tdb
Analysing hydrogen-bonding network for automated assignment of histidine
protonation. 213 donors and 184 acceptors were found.
There are 256 hydrogen bonds
Will use HISE for residue 15
8 out of 8 lines of specbond.dat converted successfully
Special Atom Distance matrix:
CYS6 MET12 HIS15 CYS30 CYS64 CYS76 CYS80
SG48 SD87 NE2118 SG238 SG513 SG601 SG630
MET12 SD87 1.167
HIS15 NE2118 1.776 1.018
CYS30 SG238 1.406 1.054 2.068
CYS64 SG513 2.835 1.793 1.789 2.241
CYS76 SG601 2.705 1.551 1.468 2.115 0.765
CYS80 SG630 2.959 1.950 1.916 2.390 0.198 0.944
CYS94 SG724 2.550 1.407 1.382 1.974 0.666 0.201 0.855
MET105 SD799 1.827 0.911 1.683 0.888 1.849 1.461 2.036
CYS115 SG889 1.577 1.084 2.078 0.200 2.111 1.989 2.262
CYS127 SG981 0.198 1.072 1.720 1.313 2.799 2.622 2.933
CYS94 MET105 CYS115
SG724 SD799 SG889
MET105 SD799 1.381
CYS115 SG889 1.853 0.790
CYS127 SG981 2.475 1.685 1.483
Linking CYS-6 SG-48 and CYS-127 SG-981...
Linking CYS-30 SG-238 and CYS-115 SG-889...
Linking CYS-64 SG-513 and CYS-80 SG-630...
Linking CYS-76 SG-601 and CYS-94 SG-724...
Making bonds...
Number of bonds was 1984, now 1984
Generating angles, dihedrals and pairs...
Before cleaning: 5142 pairs
Before cleaning: 5187 dihedrals
Keeping all generated dihedrals
Making cmap torsions...
There are 5187 dihedrals, 426 impropers, 3547 angles
5106 pairs, 1984 bonds and 0 virtual sites
Total mass 14313.193 a.m.u.
Total charge 8.000 e
Writing topologyWriting coordinate file...
:-) GROMACS - gmx editconf, 2020.3-MODIFIED (-: GROMACS is written by:Emile Apol Rossen Apostolov Paul Bauer Herman J.C. Berendsen
Par Bjelkmar Christian Blau Viacheslav Bolnykh Kevin Boyd
Aldert van Buuren Rudi van Drunen Anton Feenstra Alan Gray
Gerrit Groenhof Anca Hamuraru Vincent Hindriksen M. Eric Irrgang
Aleksei Iupinov Christoph Junghans Joe Jordan Dimitrios Karkoulis
Peter Kasson Jiri Kraus Carsten Kutzner Per Larsson
Justin A. Lemkul Viveca Lindahl Magnus Lundborg Erik Marklund
Pascal Merz Pieter Meulenhoff Teemu Murtola Szilard Pall
Sander Pronk Roland Schulz Michael Shirts Alexey Shvetsov
Alfons Sijbers Peter Tieleman Jon Vincent Teemu Virolainen
Christian Wennberg Maarten Wolf Artem Zhmurov
and the project leaders:
Mark Abraham, Berk Hess, Erik Lindahl, and David van der SpoelCopyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2019, The GROMACS development team at
Uppsala University, Stockholm University and
the Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.GROMACS is free software; you can redistribute it and/or modify it
under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.GROMACS: gmx editconf, version 2020.3-MODIFIED
Executable: C:\Users\User\AppData\Local\OneAngstrom\SAMSON-Data\1.0.0\Scratch\Elements\02407D21-0490-30BA-D20F-2E88DB100FC5\Resource\gromacs\gmx.exe
Data prefix: C:\Program Files\Gromacs
Working dir: C:\Users\User\AppData\Local\OneAngstrom\SAMSON-Data\1.0.0\Scratch\Elements\02407D21-0490-30BA-D20F-2E88DB100FC5\Resource\gromacs
Command line:
gmx editconf -f C:/Users/User/AppData/Local/OneAngstrom/SAMSON-Data/1.0.0/Scratch/GROMACS/GeneratedFiles/1AKI_model.gro -o C:/Users/User/AppData/Local/OneAngstrom/SAMSON-Data/1.0.0/Scratch/GROMACS/GeneratedFiles/1AKI_boxed.gro -c -box 4.234 -bt cubicProgram: gmx editconf, version 2020.3-MODIFIED
Source file: src\gromacs\fileio\groio.cpp (line 67)Fatal error:
gro file does not have the number of atoms on the second lineFor more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
Note that major changes are planned in future for editconf, to improve usability and utility. -
Dear Guy,
thank you for your interest in SAMSON and the GROMACS Wizard extension. Could you please tell me whether you clicked "Compute fitted box" first, to indicate what system you want to simulate? It looks like nothing was selected, and no atoms were exported. I just did a quick test with 1AKI (the PDB indicated in the tutorial):
- removed the waters using the document view
- clicked Compute fitted box
- increased the box size to 7,015nm
- checked Add solvent and Neutralize system (using Ca and Cl)
- clicked Prepare
Here is what I obtain: (I added a secondary structure in the Biology menu)
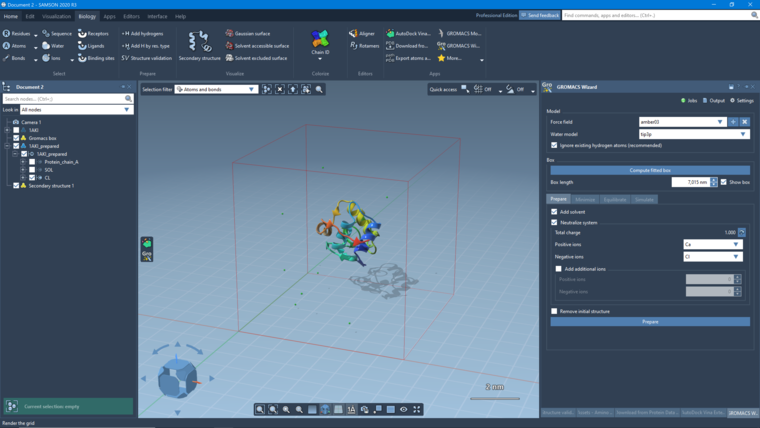
If you'd like, you can use the 1AKI tutorial adapted for the GROMACS Wizard: https://documentation.samson-connect.net/gromacs-wizard/. Please let us know if you have any questions.
thanks,
best,Stephane
-
The 1AKI pdb file was imported and cleaned of all the water molecules.
I didn't increase the sized of the box and it was a 4,115 nm box. Add solvent and Neutralize system was selected using Na and Cl. Then after clicking on prepare I got the file reported in my first post.
I used the oplsaa forcefield with the spc water model. I also give a try to your parameter (amber03 instead of oplsaa, tip3p instead of spc, and Ca instead of Na). But this lead me to the same result.
If I try to generate a gro file using the Model Generator, it generate one file but this one doesn't work for all the other calculations.
Here is my screen :
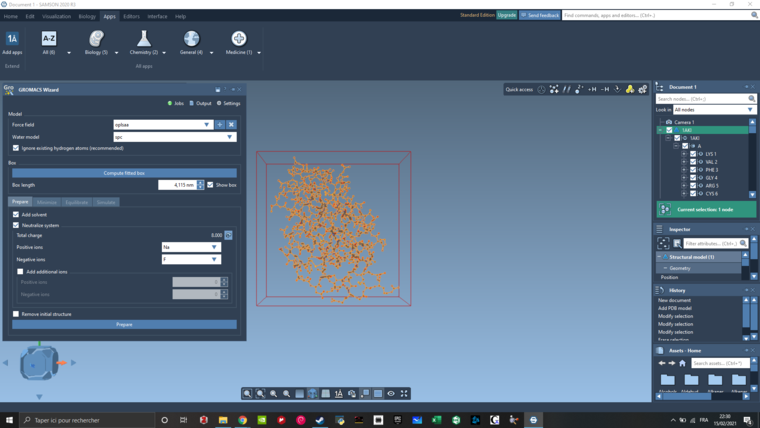
-
Could you please tell me whether you added custom force fields? (Using the + sign). If yes, please try to remove them (using the X sign) and try again, and let me know.
Thanks,
Stephane -
No, I just have the force fields provided with the extension. Even though I tried removing with the X sign, no change.
Thanks for your help -
Ok thanks. I'm going to contact you by email for further exchange.