Manually Add Hydrogen Bonds
-
Hi! Posting this as a feature request as I was not able to find a way do it. I would love to be able to customize bonds a little more: specifically, hydrogen bonds are only added via the Hydrogen Bond Finder tool, which can at times not recognize some non-covalent interactions that I would like to visualize. I imagine this could be done perhaps by manually adding a bond and adding the ability to modify its appearance to make them dashed, just like Atoms have a "Display" option to be set in the Inspector. Similarly, during Presentations when molecular fragments move, these dashed bonds could ideally behave like Hydrogen Bonds (appear only when donor and acceptor are close enough) instead of normal bonds (always show regardless of the distance).
I never looked into developing extensions for SAMSON, but if this sounds like some manageable problem and somebody is able to point me in the right direction, I would be happy to take a stab at it!
Best,
Nick -
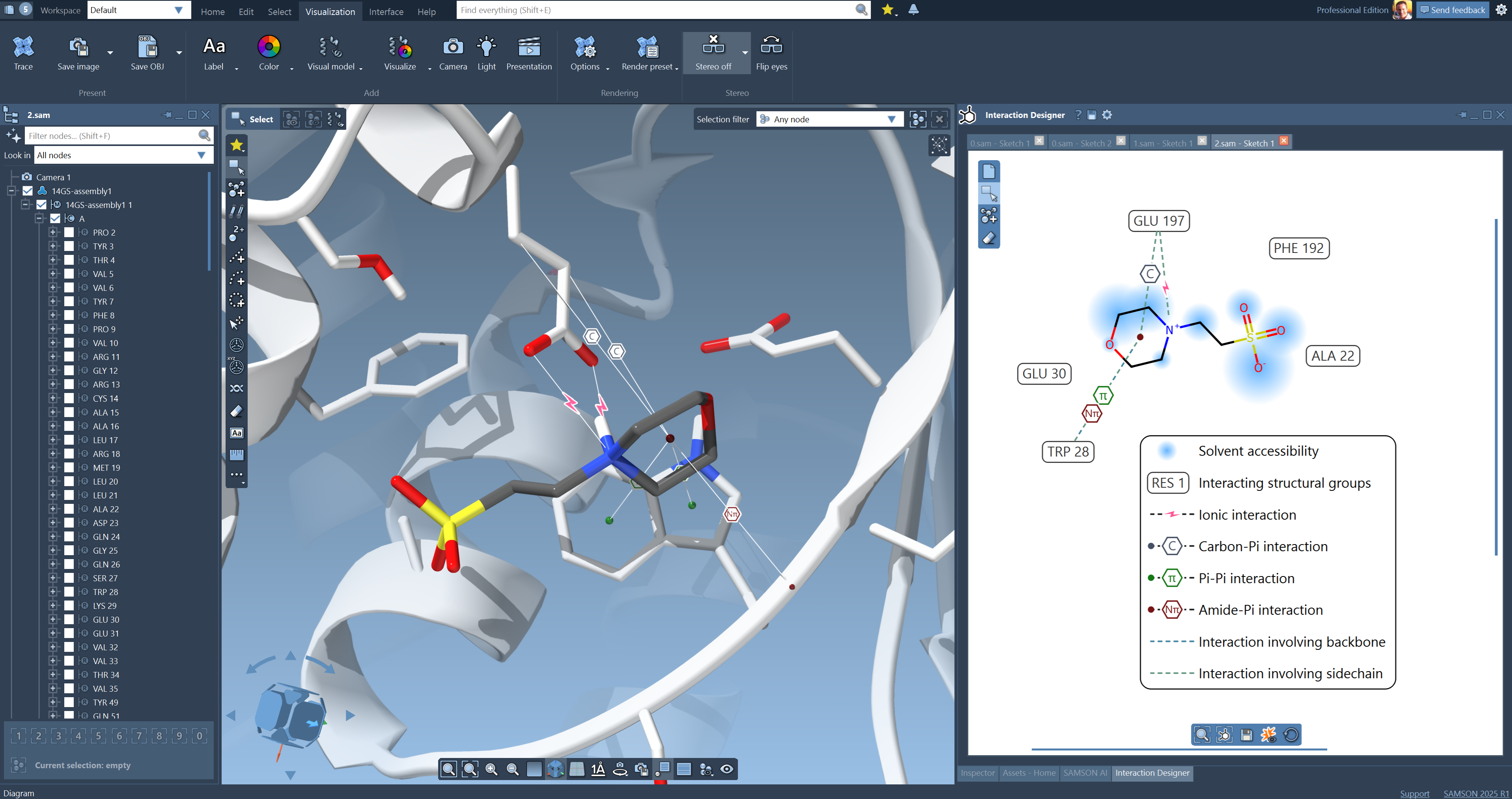
Hi Nick, the new Interaction Designer (included with SAMSON) can now show different types of interactions in the 3D viewport:
Would this work for you? You can use the Home > Diagram command to activate this. -
Hi, thank you for the swift reply! I tried the Interaction Designer but it was not quite what I was looking for. The transition state structures I am working with seem more challenging for the software, and I would like to specify/toggle each interaction myself, if possible (including potentially deleting individual hydrogen bonds that are automatically generated by the Hydrogen Bond Finder tool).
-
Hi @Nicolo-0 ,
We have exposed H-bond functionality in our Python API - now you can generate H-bonds using Python scripting in SAMSON. See SBHydrogenBondGroup, SBHydrogenBond.
You can:
- specify for which systems to generate H-bonds
- remove H-bonds
- add H-bonds
Note: H-bonds are stored within an H-bond group, which itself is stored in a structural model. On the other hand, the Hydrogen Bond Finder provides the results as just a visual model for some historical/performance reasons.
Here is a sample code to add H-bonds based on a structural model or between receptors and ligands (see Python Scripting Guide: Selecting for more information on how to get nodes):
### create H-bonds # get an indexer of all structural models in the active document structural_model_indexer = SAMSON.getNodes("node.type structuralModel") # get the first structural model structural_model = structural_model_indexer[0] # get an indexer with receptor and ligand nodes receptor_indexer = SAMSON.getNodes("node.category receptor") ligand_indexer = SAMSON.getNodes("node.category ligand") with SAMSON.holding("Add H-bonds"): # create an H-bond group object between atoms in a single node indexer h_bond_group = SBHydrogenBondGroup("H-bonds", structural_model_indexer) # create an H-bond group object between atoms in two node indexers #h_bond_group = SBHydrogenBondGroup("H-bonds between receptor and ligand", receptor_indexer, ligand_indexer) # compute H-bonds h_bond_group.update() # hold the node for undo/redo SAMSON.hold(h_bond_group) # create the H-bond group h_bond_group.create() # add the node to the active document structural_model.addChild(h_bond_group)The newly created H-bond group will be placed in the structural model - expand it in the Document view to see it. You can access its options by selecting it and opening the Inspector.
You can remove H-bonds from an H-bond group based on some criteria, e.g., based on the donor-acceptor distance:
### remove some H-bonds from the H-bond group # get H-bonds from the H-bond group, exclude the H-bond group itself hbond_indexer = h_bond_group.getNodes("not n.t hbg") print(f"The total number of computed H-bonds is {len(hbond_indexer)}") with SAMSON.holding("Remove some H-bonds"): for hbond in hbond_indexer: if hbond.donorAcceptorDistance > SBQuantity.angstrom(3.5): print(hbond) h_bond_group.removeChild(hbond) updated_hbond_indexer = h_bond_group.getNodes("not n.t hbg") print(f"The total number of computed H-bonds is {len(updated_hbond_indexer)}")You can also add new H-bonds in a H-bond group, as follows:
### add an H-bond in the H-bond group with SAMSON.holding("Add new H-bonds"): # create an H-bond object based on already defined acceptor, donor, and (optionally) hydrogen atoms hbond = SBHydrogenBond(acceptor, donor) #, hydrogen # hold the node for undo/redo SAMSON.hold(hbond) # create the H-bond hbond.create() h_bond_group.addChild(hbond)Let me know if you have any questions or require some help.
-
@DmitriyMarin Hi Dmitriy, thanks a lot, this looks amazing! I will look into it and let you know if I have any questions.
My best,
Nick -
@DmitriyMarin Would it be possible to modify the "Direction" attribute just like the previous Hydrogen Bond Finder structural model? (i.e. showing the HBs from Hydrogen to acceptor). I could not find it in the Inspector panel for the new "H-bonds" structural model, but maybe from the console?
-
Hi @Nicolo-0 ,
No, currently, there is no option to specify that H-bonds in an H-bond group (which is part of a structural model) should be displayed between hydrogens and acceptors - they are drawn between donors and acceptors, since hydrogens might be implicit. However, we can add such an option in the next major release of SAMSON.
But if you want it just for the sake of visualization, then you can try setting hydrogens as donors programatically (via Python). Note that this should be done without callingupdate()since this will recompute the bonds.